Research
The Pharmaceutical Bioinformatics lab is working on several projects dealing with the discovery of new drugs, analysis of the mechanisms of known compounds as well as the biosynthesis of natural compounds. Some selected projects are described here.
Automated recognition of functional compound-protein relationships in literature

PubMed is a database containing millions of references to biomedical publications. Searching for protein-compound interactions in this continuously growing amount of literature can be a difficult and time-consuming task.
Text mining and machine learning techniques are applied here to identify the functional compound-protein relationships in all titles and abstracts of the PubMed database. (Döring et al., 2020)
Computational Pathology - Medical Image classification
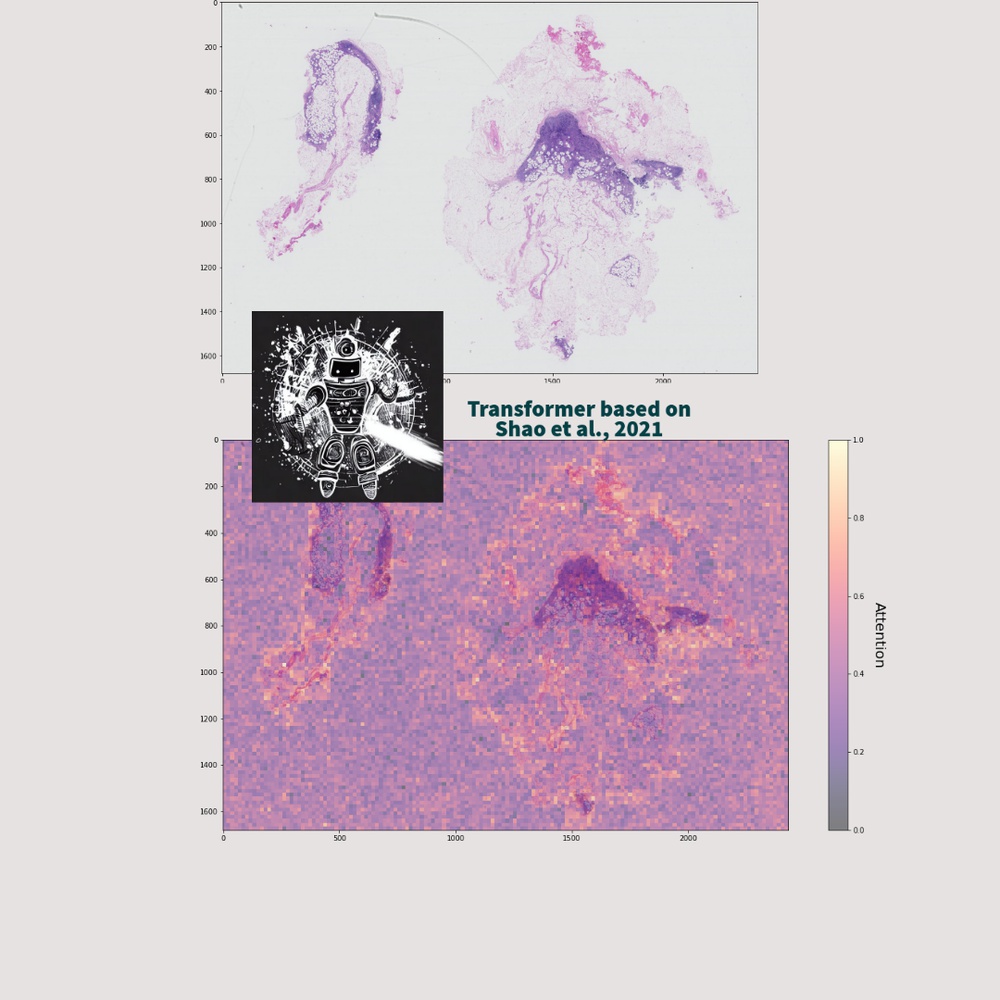
Computational pathology and medical image classification contribute to the field of healthcare by leveraging machine and deep learning technologies to analyze and interpret medical images, leading to advancements in the automation of tasks such as disease detection, prognosis, and treatment planning.
In particular, the analysis of high-dimensional data present in whole slide images (WSIs) has become a focal point for researchers and practitioners in the field. These images are exceptionally large and contain billions of pixels, resulting in enormous amounts of high-dimensional data.
AI models, leveraging the power of the Transformer or CNN structure, can effectively capture subtle morphological variations and spatial relationships within WSIs, thereby enabling precise identification of abnormalities or disease markers.
We are focused on the application and development of methods in a realistic setting of highly diverse and messy data, while retaining a high quality prediction over a broad range of skin diseases.
Epigenetic drug discovery
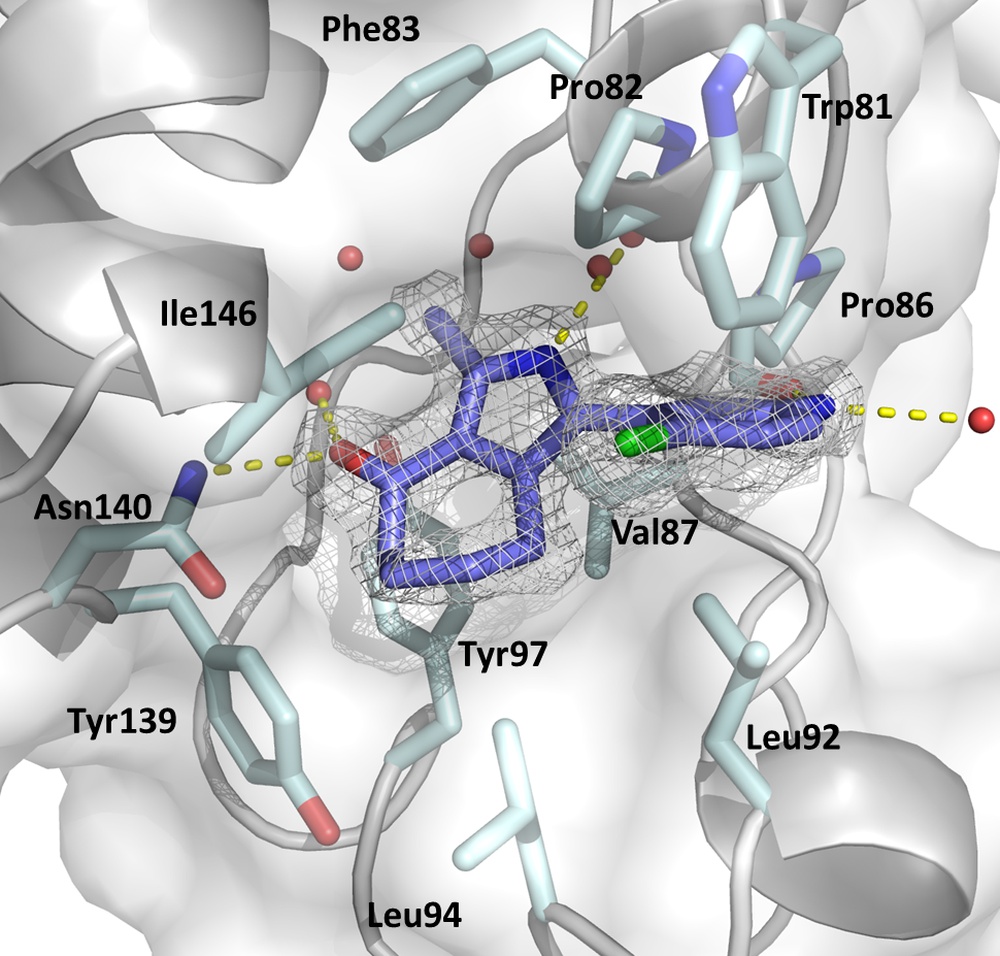
Epigenetic mechanisms are essential for normal cellular development and maintenance of cellular homeostasis of all eukaryotes. In the past few years, it has been well-established that epigenetic aberrations play an important role in a wide range of human diseases. Until now, the focus of research has been on cancer. In recent years, promising new therapeutic methods and drugs have been developed that are currently in clinical trials or have already received FDA approval. More recent research is trying to extend this success story to diseases caused by eukaryotic parasites, such as malaria, leishmaniasis or trypanosomiasis. For this purpose, either new inhibitors can be designed or inhibitors of human homologs can be repurposed.
We use in silico methods (including molecular docking and virtual screening as well as ligand-based approaches such as pharmacophore modeling and QSAR, network analysis and MD simulations) for the design of inhibitors for a broad array of potential targets including epigenetic targets (e.g. bromodomains, chromodomains, HDACs etc.), methyltransferases as well as cofactor-binding proteins. To verify our predictions, we produce target proteins by heterologous expression, which after purification are used for affinity measurement (ITC) and structure elucidation (X-ray crystallography). In addition, we have access to synthesis and in vitro testing capabilities through our collaborators.
The figure displays the KAc binding sites of BRD4(1) in complex with the pan-selective bromodomain Inhibitor MPM6 (Warstat et al., 2023).
Genome-based secondary metabolite prediction
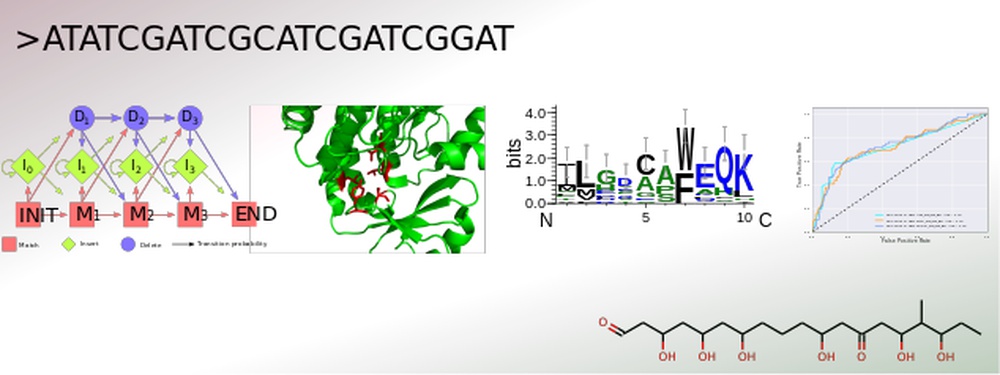
The secondary metabolism of bacteria, fungi and plants yields a vast number of bioactive substances. The constantly increasing amount of published genomic data provides the opportunity for an efficient identification of gene clusters by genome mining. Conversely, for many natural products with resolved structures, the encoding gene clusters have not yet been identified. Structural elucidation of the actual secondary metabolite is still challenging, especially due to the currently unpredictable post-modifications.
To address this, SeMPI was designed, a web server providing a Secondary Metabolite Prediction and Identification pipeline for natural products synthesized by polyketide synthases of type I modular (Zierep et al., 2017). Further extensions of SeMPI require the improvement of state-of-the-art prediction algorithms, but also the implementation of new algorithms adapted to the metabolite in focus.
The core of all secondary metabolite prediction approaches is based on the accurate functional classification of the proteins responsible for its synthesis. In order to facilitate this task a pipeline was designed which merges all crucial steps for efficient protein classification. It allows for the collection and annotation of related sequences. These can be used for parallel benchmarking of suitable machine learning algorithms, such as hidden Markov profiles, position specific scoring matrices and optimized decision trees, accompanied by careful parameter optimization of each design. The most efficient classification system can then be incorporated into the prediction software. Advantages of this set-up are the straightforward evaluation of a newly created classification algorithm, as well as an update option, which keeps the learning data up-to-date and therefore the ability to build prediction rules for various kinds of gene cluster products, such as polyketides of type I iterative and nonribosomal peptides.
Currently the feasibility of structure based classifications are also evaluated by incorporation of secondary structure assignment tools.
Molecular Dynamics (MD) simulation of therapeutic relevant protein targets
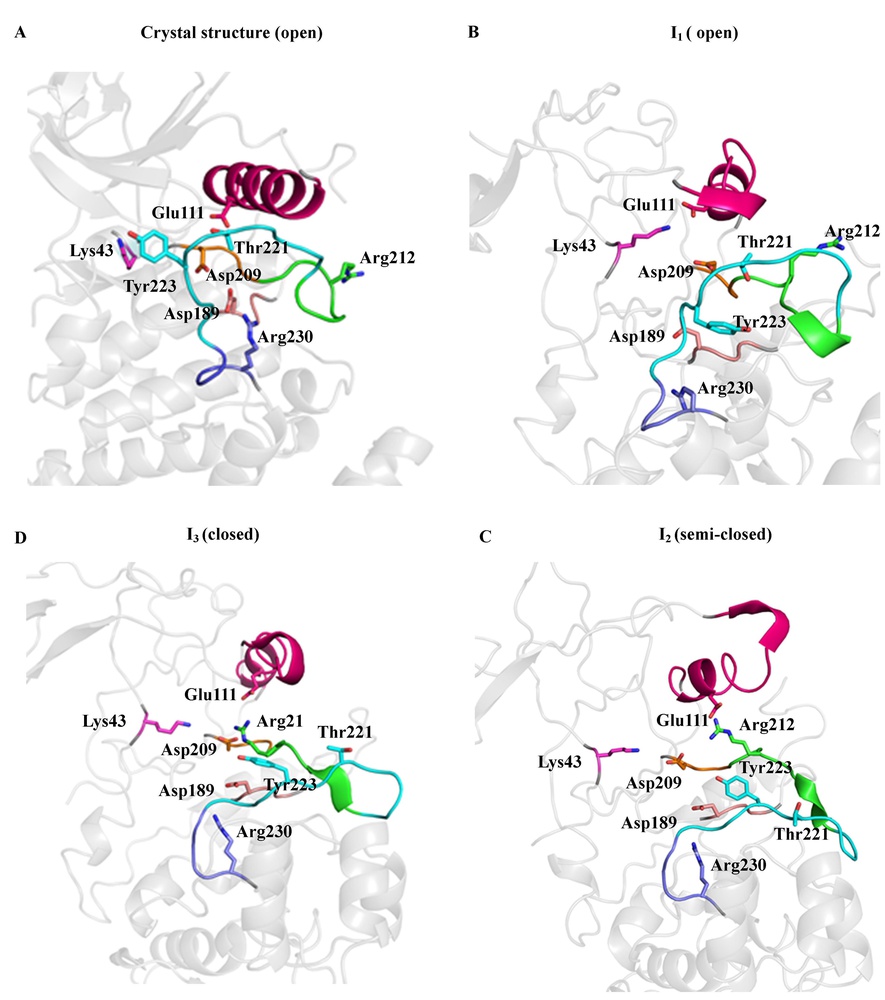 We are interested in understanding the structural dynamics and the signaling cascade mechanism of relevant biological targets with therapeutic potentials. We study the microscale atomistic dynamics through distributed MD simulations, using a BinAC high performance computing cluster. Currently, we are working on kinases and epigenetic drug targets. (Publication in preparation.)
We are interested in understanding the structural dynamics and the signaling cascade mechanism of relevant biological targets with therapeutic potentials. We study the microscale atomistic dynamics through distributed MD simulations, using a BinAC high performance computing cluster. Currently, we are working on kinases and epigenetic drug targets. (Publication in preparation.)
Structural Analysis of Privileged Chemotypes in Drug Discovery
 CovPDB is a freely accessible web database solely dedicated to high-resolution 3D structures of biologically relevant covalent protein-ligand complexes, mined from the Protein Data Bank (PDB). We have so created CovPDB to assist structure-based approaches in chemical biology and drug design by identifying covalent binding sites suitable for the docking of drug-like ligands, and, likewise, typical ligands that covalently modify targetable binding sites. Furthermore, the corresponding covalent bonding mechanisms of such complexes were manually and expertly annotated. For these curated complexes, the chemical structures and warheads of pre-reactive electrophilic ligands as well as the covalent bonding mechanisms to their target proteins were expertly manually annotated. Totally, CovPDB contains 733 proteins and 1,501 ligands, relating to 2,294 cP-L complexes, 93 reactive warheads, 14 targetable residues, and 21 covalent mechanisms. Users are provided with an intuitive and interactive web interface that allows multiple search and browsing options to explore the covalent interactome at a molecular level in order to develop novel TCIs (Gao et al., 2021).
Based on a comprehensive characterization of the covalently targeted cysteine residues, we developed a machine learning model covalent cysteine predictor (HyperCys) to identify targeted cysteine residues (Gao & Günther, 2023).
CovPDB is a freely accessible web database solely dedicated to high-resolution 3D structures of biologically relevant covalent protein-ligand complexes, mined from the Protein Data Bank (PDB). We have so created CovPDB to assist structure-based approaches in chemical biology and drug design by identifying covalent binding sites suitable for the docking of drug-like ligands, and, likewise, typical ligands that covalently modify targetable binding sites. Furthermore, the corresponding covalent bonding mechanisms of such complexes were manually and expertly annotated. For these curated complexes, the chemical structures and warheads of pre-reactive electrophilic ligands as well as the covalent bonding mechanisms to their target proteins were expertly manually annotated. Totally, CovPDB contains 733 proteins and 1,501 ligands, relating to 2,294 cP-L complexes, 93 reactive warheads, 14 targetable residues, and 21 covalent mechanisms. Users are provided with an intuitive and interactive web interface that allows multiple search and browsing options to explore the covalent interactome at a molecular level in order to develop novel TCIs (Gao et al., 2021).
Based on a comprehensive characterization of the covalently targeted cysteine residues, we developed a machine learning model covalent cysteine predictor (HyperCys) to identify targeted cysteine residues (Gao & Günther, 2023).